
Drug development requires an average overall investment of USD 1.5 – 4.5 billion and remains at high risk due to the huge attrition rate phase after phase. The pre-clinical development phase and the phase 1 clinical trial show the highest attrition rate due to toxicity or drug metabolism and pharmacokinetic issues. Mitigating the risk during a new drug candidate development program starting from the discovery phase onwards remains essential for drug development efficiency and success.
The challenge is to perform the necessary steps in a structured and predictive manner which is also tailored to the project at hand. This can be achieved by committing the resources to stage-appropriate steps and confirming their suitability for the following steps.
This structured and stepwise project plan reduces the level of investments into future work prior to receiving positive results from the earlier stage which would justify such investments.
Doing the right things at the right time
Drug development requires an average overall investment of USD 1.5 – 4.5 billion and remains at high risk due to the huge attrition rate phase after phase [1; 2]. The pre-clinical development phase and the phase 1 clinical trial show the highest attrition rate due to toxicity or drug metabolism and pharmacokinetic issues [3; 4]. Mitigating the risk during a new drug candidate development program starting from the discovery phase onwards remains essential for drug development efficiency and success.
The challenge is to perform the necessary steps in a structured and predictive manner which is also tailored to the project at hand. This can be achieved by committing the resources to stage-appropriate steps and confirming their suitability for the following steps. This structured and stepwise project plan reduces the level of investments into future work prior to receiving positive results from the earlier stage which would justify such investments.
For small molecules and biologics, development of the compound synthesis including analytical development is the starting point into the pre-clinical and clinical program. Drug synthesis for toxicology and animal studies is a critical development step as it also serves the clinical trial program. The evaluation of toxicity depends on the chemical structure of the active compound and the purity achieved in the drug synthesis. The route of synthesis, the raw materials and solvents used, as well as the process conditions applied are important factors for the potential formation of harmful impurities. To assure the validity of the outcomes of the toxicology study, a robust and efficient drug synthesis including reliable analytical procedures is essential as this provides the necessary consistency throughout the program.
Developing a robust drug substance synthesis
Setting the compound profile targets and performing an intensive risk assessment using a structured approach, as well as prior knowledge and compound synthesis expertise ensure that the resulting material fulfills the defined quality criteria. Developing the drug substance synthesis has to be accompanied by a thorough understanding of the physicochemical properties and their relevance for drug development [5]. For example, chemical structures that are prone to drug instability or degradation, residual solvents, or formation of chemical artefacts might have a negative impact on the toxicology profile that would be preventable [6]. Other critical factors like salt selection, druggability, solubility, BA, or potential risk for polymorphs, etc. might also arise from the risk assessment and can be addressed. The expected outcome is the definition of potential Critical Material Attributes (CMA) and Critical Quality Attributes (CQA), summarized in preliminary/target specifications, at early stages in order to prevent issues with the compound and its synthesis [7; 8]. Drug substance synthesis must therefore be accompanied by scientifically sound, product-specific analytical procedures for the characterization of the active and residual byproducts. The analytical methods will establish a comprehensive drug substance profile which will be the benchmark for the clinical and commercial phase.
Developing phase-appropriate analytical methods for the active compound
The analytical method development during early-stage drug development is an evolving process that builds successively on sound scientific approaches and data as described by the FDA (Box 1).
| 21 CFR 312.23 Investigational new drug application (IND)
7 (i) … FDA recognizes that modifications to the method of preparation of the new drug substance and dosage form and changes in the dosage form itself are likely as the investigation progresses. Therefore, the emphasis in an initial Phase 1 submission should generally be placed on the identification and control of the raw materials and the new drug substance. Final specifications for the drug substance and drug product are not expected until the end of the investigational process. 7 (iv) (a ) Drug substance. A description of the drug substance, including its physical, chemical, or biological characteristics; the name and address of its manufacturer; the general method of preparation of the drug substance; the acceptable limits and analytical methods used to assure the identity, strength, quality, and purity of the drug substance; and information sufficient to support stability of the drug substance during the toxicological studies and the planned clinical studies. Reference to the current edition of the United States Pharmacopeia – National Formulary may satisfy relevant requirements in this paragraph. |
Box 1: Requirements for drug substance [15]
Regulatory authorities consider this as phase or stage-appropriate analytical method development. They expect only the full analytical method validation according to ICH guidelines to be provided along with the final drug product presentation entering into phase 3 clinical trials. During the pre-clinical and IND phase regulatory authorities request that “an analytical procedure is to demonstrate that it is suitable for its intended purpose” [9]. This is further specified in the ICH Q7 guideline by stating that “while analytical methods performed to evaluate a batch of API for clinical trials may not yet be validated, they should be scientifically sound.” [10]. The selection and development of a scientifically sound analytical method requires experience and understanding of a method principle and capability to measure the targeted CMQ or CQA reliably. For example, to determine elemental impurities Inductively Coupled Plasma Mass Spectroscopy (ICP-MS) or for residual solvents
Head-space Gas Chromatography (HSGC) are suitable method principles. The suitability for the method’s intended purpose is often established by validation of a subset of ICH validation parameters.
The analytical development for a drug substance starts with a clear definition of the targeted drug substance profile and the limits of acceptability (table 1). Analytical methods have to be scientifically sound and fit for purpose to accurately measure the target in the specific drug compound.

Table 1: Ardena’s targeted drug substance profile of quality attributes, acceptance criteria and potential analytical procedures for GLP toxicology study use
(*) In this early phase a well-characterized analytical reference standard may not be available, hence assay by HPLC cannot be used.
Phase-appropriate analytical method development in the scope of product development
The pharmaceutical development process consists of a matrix of different work steps, investigations, and documentation, all of which are interrelated and build on each other (Figure 1). A successful development plan for a new drug product consists of a systematic and knowledge-based approach. This is especially true for the synthesis development of the drug substance, which enables the production of a highly pure drug substance in a reproducible and scalable manner. During clinical stages analytical methods for qualification of the raw materials and intermediates of the synthesis are established as well. Critical solvents and process parameters should be kept to a minimum. The ultimate objective is to manufacture the drug substance by the same synthetic route, and purification and crystallization processes from the toxicology supplies through to the market. In practice however, continuous improvements are often encountered up to the start of process validation. From the beginning the critical equipment and ancillary systems used in manufacturing and control must be suitable and qualified for the intended purpose. To streamline the development times and prevent unnecessary stringent API quality (and its associated cost), Ardena uses the demonstration batch of the drug substance as the first GLP batch to cover the supplies for the toxicology studies within the predefined impurity targets. The early provision of the drug substance synthesis provides continuity within the drug development program, leverages important prior product and process knowledge, and reinforces product specific learning according to the ICH Q 11 recommendations [6].

Figure 1: The pharmaceutical drug product and analytical method development process matrix
Early-stage analytical methods development along with the GLP drug substance batch manufacturing will be defined and selected based on a mechanistic understanding of the method principles, internal expertise, and data from prior experiments in accordance with the FDA guidelines [11; 12]. This also ensures that the analytical methods developed are meaningful and can be validated later during the GMP drug substance and drug product manufacturing if the toxicology studies support the nomination of the molecule for further clinical studies. Parameters that can be considered and may be evaluated during analytical method development are specificity, linearity, limits of detection (LOD), limits of quantitation (LOQ), range, accuracy, and precision [10]. The analytical method development in early stage starts with a clearly defined targeted drug substance profile including the quality attributes and acceptance criteria derived from the risk assessment (Table 1). For the toxicology study the goal of the analytical method development is to select the right procedure to determine the individual targets qualitatively and quantitatively with sufficient reliability. This includes the detection of predictable and unpredictable impurities and their characterization [13]. Each individual step and every result must be precisely documented, and corresponding samples have to be taken for later validation purposes. It should be noted that the suitability of the method as well as important validation parameters are revisited and justified during the course of the clinical program based on increasing learning and evidence, and possible changes in the manufacturing process. It is not uncommon for an analytical validation parameter to become more stringent as development progresses for the drug compound and its finished dosage form. The phase-appropriate approach for the process as well as the analytical development is summarized in figure 2.
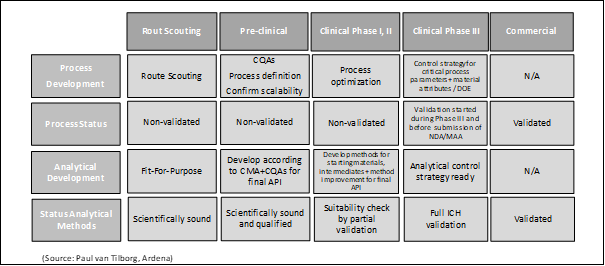
Figure 2: Requirements for process and analytical method development using a phase-appropriate approach
The early development of the synthetic route of a drug substance and the corresponding analytical methods must be continuously documented according to the CMC Guidelines which form an important part of the evaluation of the toxicology studies, the IND submissions for the clinical phases, and finally the drug product approval [14].
Conclusion
Both the synthesis of a drug substance and its analytical methods should be developed and defined early on in a scientifically sound and data-based manner, as these are the foundation for meaningful toxicological evaluation and sufficient patient safety throughout the clinical phase.
Ardena applies phase-appropriate criteria for the qualification/validation of analytical methods during development. The key parameters taken into account are those referred to in ICH Q2 (R1): specificity, linearity, precision, accuracy, and LOD/LOQ. While the equipment used must be qualified from the outset, the full ICH validation of the process and analytical methods only takes place during the clinical development process. An efficient project plan combining drug substance synthesis and analytical method development through phase-appropriate deliverables along the development process ensures efficient pharmaceutical development and regulatory compliance.
References
- Schlander et al (2021) How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. PharmacoEconomics 39:1243–1269
- Wouters et al (2020) Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA ;323(9):844-853
- Waring MJ et al (2015) An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nature Rev Drug Discov 14:475-486
- Roberts RA et al (2014) Reducing attrition in drug development: smart loading preclinical safety assessment. Drug Discov Today 19(3):341-347
- Brown DG & Boström J (2016) Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? Med. Chem. 2016, 59, 4443-4458
- ICH Q11 (2012) Development and manufacturing of drug substances (chemical entities and biological entities). https://database.ich.org/sites/default/files/Q11%20Guideline.pdf
- Taylor CR et al (2020) Minimizing Polymorphic Risk through Cooperative Computational and Experimental Exploration. J. Am. Chem. Soc. 142, 16668-16680
- Black SN et al (2007) Structure, Solubility, Screening, and Synthesis of Molecular Salts. J Pharm Sci 96:1053-1068
- 21 CFR 312.23 Investigational new drug application (IND) from Oct 1, 2021 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=312.23
- ICH Q2(R1) (2005) Validation of analytical procedures: text and methodology. https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf
- ICH Q 7 (2020) Good manufacturing practice guide for active pharmaceutical ingredients. https://database.ich.org/sites/default/files/Q7%20Guideline.pdf
- FDA (2015) Analytical Procedures and Methods Validation for Drugs and Biologics. July 2015. https://www.fda.gov/files/drugs/published/Analytical-Procedures-and-Methods-Validation-for-Drugs-and-Biologics.pdf
- ICH Q3A (R2) (2006) Impurities in new drug substances. https://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf
- FDA CMC Guidances web site: https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/cmc-and-gmp-guidances
- 21 CFR 312.23 Investigational new drug application (IND) from Oct 1, 2021 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=312.23
